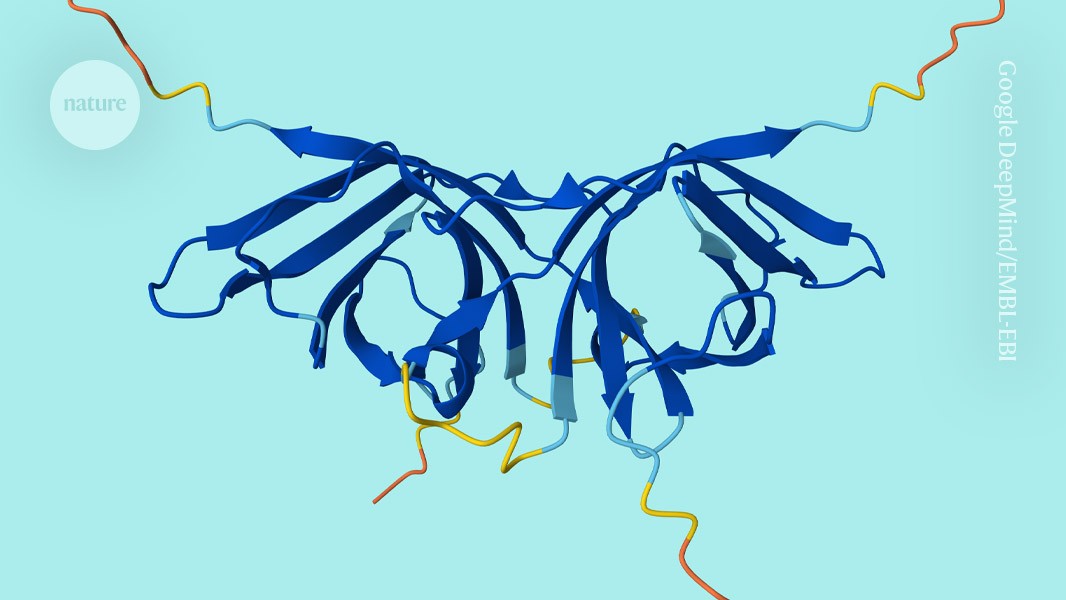
"Since its release in 2021, this repository has become a bedrock in discovery and a first port of call for research projects that try to understand life at the molecular level. But previous iterations of the database lacked predictions of how proteins form complexes, which can be indispensable for their function."
"For instance, HIV-1 protease - a viral protein that is a key drug target - works only when two copies of the same protein form a working enzyme. Such proteins were already included in the database as individual 'monomers' but their entries tell only part of their story."
"To make predictions for even small complexes of two proteins was a crucial challenge, says Steinegger. 'It is quite a different beast than monomer predictions.' Protein-complex predictions are exceedingly intensive computationally, so a consortium - including Steinegger's lab, EMBL-EBI, Google DeepMind and chipmaker NVIDIA - was formed to take on the effort."
The AlphaFold protein-structure database has expanded to include predictions of protein complexes, specifically adding 1.7 million homodimers—pairs of identical interacting protein molecules. Previously containing around 200 million predictions of individual protein structures generated by Google DeepMind's AlphaFold2 AI tool, the database now captures how proteins function together. This expansion addresses a critical gap, as many proteins require complex formation to work properly, such as HIV-1 protease which functions only when two identical copies combine. The freely available database, maintained by EMBL-EBI in the UK, has become essential for molecular-level research since its 2021 release. A consortium including computational biologists, EMBL-EBI, Google DeepMind, and NVIDIA collaborated to overcome the computational intensity of predicting protein complexes.
#alphafold-database-expansion #protein-complexes #homodimers #structural-biology #ai-protein-prediction
Read at Nature
Unable to calculate read time
Collection
[
|
...
]